Consensus sulla gestione diagnostico-terapeutica della Sindrome di Nelson
Adriana Albani1, Erika Messina2
1Department of Endocrinology, Medizinische Klinik und Poliklinik IV, LMU Klinikum, Ludwig Maximilians University Munich, München, Germany; 2Istituto Clinico Mater Domini, Castellanza, Italia Autore corrispondente: Adriana Albani
[email protected]
Introduzione
La Sindrome di Nelson è una possibile complicazione dopo surrenectomia bilaterale per malattia di Cushing, storicamente definita dalla presenza di tre criteri, ovvero progressione di un tumore ipofisario ACTH-secernente, iperpigmentazione ed elevati livelli di ACTH [1].
Recentemente un gruppo di esperti nel campo della patologia ipotalamo-ipofisaria ha rivisitato e discusso la letteratura sulla sindrome di Nelson, emanando delle nuove raccomandazioni [2] La prima raccomandazione si incentra sulla terminologia della sindrome. In accordo con quanto proposto nel 2007 [3] si suggerisce di sostituire il termine “sindrome di Nelson” con “Progressione del tumore corticotropo dopo surrenectomia bilaterale/sindrome di Nelson”, abbreviato CTPBADX/NS. La ragione di questa modifica risiede nel focalizzare l’attenzione verso la caratteristica chiave della sindrome, ovvero la progressione tumorale.
Contestualmente i criteri necessari per la diagnosi di CTP-BADX/NS vengono rivisitati, suggerendo come criterio principale l’evidenza radiologica di progressione tumorale o di comparsa di tumore ipofisario dopo surrenectomia bilaterale. La presenza di iperpigmentazione cutanea e un incremento dei valori di ACTH dopo surrenectomia bilaterale, dotati di bassa specificità e sensibilità, vengono considerati criteri secondari non necessari per la diagnosi [2].
Epidemiologia e diagnosi
I dati relativi all’incidenza di CTP-BADX/NS e al suo tempo medio di comparsa sono molto eterogenei tra loro. Studi relativamente recenti, che utilizzano le immagini TAC o MRT come criterio per la diagnosi riportano una prevalenza media del 43% e un intervallo di tempo medio di 2.5 anni [3-6]
Fattori predittivi dello sviluppo di CTP-BADX/NS sembrano essere elevati livelli di ACTH dopo surrenectomia [3, 7-10], evidenza radiologica di tumore ipofisario antecedente alla surrenectomia [9, 11] e l’età giovanile [12-14].
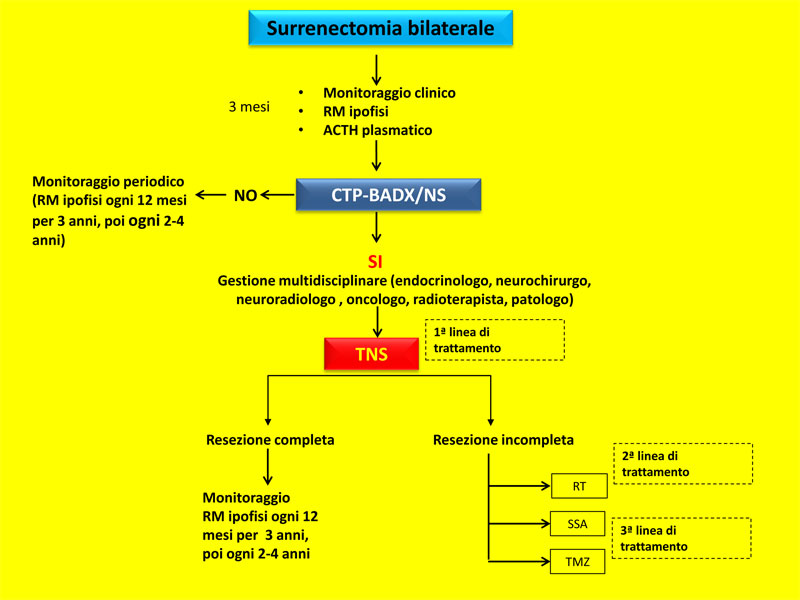
Un attento follow-up viene pertanto raccomandato particolarmente in queste tre categorie [2]. Si raccomanda l’esecuzione di una MRT tre mesi dopo l’intervento e una volta l’anno per i primi tre anni. I controlli radiologici possono essere successivamente dilazionati ogni 2-4 anni, in relazione alla clinica e ai valori di ACTH [2].
Terapia neurochirirgica e radioterapica
Una volta posta diagnosi di CTP-BADX/NS, la terapia di prima scelta raccomandata è la terapia chirurgica. In passato, a causa dei risultati non ottimali della rimozione selettiva del tumore e dell’aggressività di CTP-BADX/NS veniva praticata preferenzialmente la ipofisectomia totale [15, 16].
Col tempo grazie al perfezionamento delle tecniche chirurgiche la terapia di prima scelta è divenuta la rimozione selettiva del tumore ipofisario.
L’approccio transsfenoidale rappresenta la tecnica raccomandata, da eseguire possibilmente prima che il tumore raggiunga una estensione extrasellare [2]. La chirurgia transcranica va considerata solo in casi eccezionali, quando l’approccio transsfenoidale non è eseguibile [2].
Qualora la resezione chirurgica risulti incompleta, specialmente in presenza di un macroadenoma invasivo, può essere preso in considerazione il trattamento radioterapico concomitante, la cui indicazione va stabilita caso per caso nell’ambito di una discussione multidisciplinare.
La radioterapia è uno strumento utilizzato da tempo nella gestione della CTP-BADX/NS. In passato è stata ampiamente adoperata la radioterapia convenzionale frazionata (dose cumulativa 30-50 Gy) con risultati non sempre facilmente interpretabili, soprattutto per la mancanza di dati sugli outcome radiologici e gli effetti a lungo termine.
Più recentemente l’utilizzo delle tecniche di radiochirurgia stereotassica mediante gamma-knife (GKS) ha aumentato a >90% l’efficacia della terapia radiante nel controllo della crescita tumorale, sebbene l’incidenza cumulativa di CTP-BADX/NS dopo il trattamento GKS risulti ancora estremamente variabile [17, 18]. In generale, i tumori di dimensioni più piccole sono gestibili mediante radiochirurgia stereotassica mentre per quelli di dimensioni maggiori sembrerebbe più adatto l’utilizzo della radioterapia convenzionale frazionata.
Le raccomandazioni sono dunque quelle di trattare la CTP-BADX/NS mediante radioterapia nei casi in cui l’approccio chirurgico risulti troppo rischioso o comporti una incompleta resezione del tumore [2], mentre non vi è indicazione all’utilizzo profilattico della radioterapia per prevenire la progressione del tumore adenocorticotropo.
Tentativi di terapia medica
Non esiste una terapia medica standardizzata per la CTP-BADX/NS. Alcuni studi che hanno indagato l'effetto della terapia farmacologica utilizzando l’ACTH plasmatico come marker surrogato di crescita tumorale, non hanno difatti dimostrato prove coerenti e soddisfacenti di efficacia, né per il fattore di inibizione del rilascio di MSH (MIF) né per il rosiglitazone la ciproeptadina, il sodio valproato e gli agonisti della dopamina bromocriptina e cabergolina [2]. La temozolomide (TMZ) somministrata a pazienti con CTP-BADX/NS invasivo ha finora mostrato una efficacia molto variabile: ha prodotto la riduzione della massa tumorale e dei livelli di ACTH associati ad un miglioramento clinico in alcuni casi [19], ma è stata riportata una nuova progressione tumorale dopo 6 mesi dalla sospensione della terapia [20], o anche una risposta assente pur in mancanza di noti fattori di resistenza alla TMZ, quale la espressione della metilguanina- metiltransferasi (MGMT) [21, 22]. Una risposta biochimica e radiologica può essere ottenuta anche mediante l’utilizzo di analoghi della somatostatina di prima generazione (octreotide) [23, 24]. Il pasireotide dimostra invece una efficacia controversa sulla crescita tumorale a fronte di una riduzione media dei livelli di ACTH plasmatico del 50% [25]. Considerando l’eterogeneità clinica della CTP-BADX/NS e l’assenza di dati forti sulla efficacia delle terapie farmacologiche sembra utile, per una più appropriata scelta terapeutica, la valutazione di specifici fattori che possano avere una valenza prognostica. La presenza di mutazioni di USP8 potrebbe essere predittiva, ad esempio, di una risposta favorevole al pasireotide, mentre nei tumori corticotropi aggressivi senza mutazioni e resistenti ad altre opzioni di trattamento, viene suggerito l'uso di temozolomide [26].
Conclusioni
In conclusione, nei pazienti sottoposti a surrenectomia bilaterale viene raccomandato un primo follow-up mediante RMN 3 mesi dopo l’intervento e ogni 12 mesi per i primi 3 anni.
Successivamente i follow-up radiologici possono essere eseguiti ogni 2-4 anni in relazione ai parametri clinici e biochimici del singolo caso. La terapia di prima linea in presenza di CTP-BADX/NS è l’asportazione selettiva del tumore ipofisario, da eseguire prima che il tumore raggiunga una espansione extra sellare. La radioterapia convenzionale o la radiochirurgia vengono consigliate come terapia di seconda linea in presenza di residuo tumorale con espansione extrasellare. Il ruolo della terapia medica rimane limitato (Fig.1).
 Conflitti di interesse
Conflitti di interesse Le autrici dichiarano di non avere conflitti di interesse
Consenso informato Lo studio presentato in questo articolo non ha richiesto sperimentazione
umana
Studi sugli animali Le autrici non hanno eseguito studi sugli animali
Riferimenti bibliografici
- Nelson, D.H., et al., ACTH-producing tumor of the pituitary gland. N Engl J Med, 1958.
259(4): p. 161-4
- Reincke, M., et al., Corticotroph tumor progression after bilateral adrenalectomy (Nelson's
syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol,
2021. 184(3): p. P1-P16.
- Assie, G., et al., Corticotroph tumor progression after adrenalectomy in Cushing's Disease: A reappraisal of Nelson's Syndrome. J Clin Endocrinol Metab, 2007. 92(1): p. 172-9.
- Gil-Cardenas, A., et al., Nelson's syndrome after bilateral adrenalectomy for Cushing's disease. Surgery, 2007. 141(2): p. 147-51; discussion 151-2.
- Prajapati, O.P., et al., Bilateral adrenalectomy for Cushing's syndrome: Pros and cons. Indian J Endocrinol Metab, 2015. 19(6): p. 834-40.
- Graffeo, C.S., et al., Characterizing and predicting the Nelson-Salassa syndrome. J Neurosurg, 2017. 127(6): p. 1277-1287.
- Barnett, A.H., et al., Comparison of preoperative and postoperative ACTH concentrations after bilateral adrenalectomy in Cushing's disease. Clin Endocrinol (Oxf), 1983. 18(3): p. 301-5.
- McCance, D.R., et al., Bilateral adrenalectomy: low mortality and morbidity in Cushing's disease. Clin Endocrinol (Oxf), 1993. 39(3): p. 315-21.
- Pereira, M.A., et al., A study of patients with Nelson's syndrome. Clin Endocrinol (Oxf), 1998. 49(4): p. 533-9.
- Cohen, A.C., et al., Long-term outcome after bilateral adrenalectomy in Cushing's disease
with focus on Nelson's syndrome. Arch Endocrinol Metab, 2019. 63(5): p. 470-477.
- Sonino, N., et al., Risk factors and long-term outcome in pituitary-dependent Cushing's disease. J Clin Endocrinol Metab, 1996. 81(7): p. 2647-52.
- Hopwood, N.J. and F.M. Kenny, Incidence of Nelson's syndrome after adrenalectomy for Cushing's disease in children: results of a nationwide survey. Am J Dis Child, 1977. 131(12): p. 1353-6.
- McArthur, R.G., A.B. Hayles, and R.M. Salassa, Childhood Cushing disease: results of bilateral adrenalectomy. J Pediatr, 1979. 95(2): p. 214-9.
- Thomas, C.G., Jr., et al., Nelson's syndrome after Cushing's disease in childhood: a continuing problem. Surgery, 1984. 96(6): p. 1067-77.
- Ludecke, D., et al., Selective removal of hypersecreting pituitary adenomas? An analysis of endocrine function, operative and microscopical findings in 101 cases. Acta Neurochir (Wien), 1976. 35(1-3): p. 27-42.
- Wilson, C.B., et al., Cushing's disease and Nelson's syndrome. Clin Neurosurg, 1980. 27: p. 19-30.
- Littley, M.D., et al., Long-term follow-up of low-dose external pituitary irradiation for Cushing's disease. Clin Endocrinol (Oxf), 1990. 33(4): p. 445-55.
- Tran, L.M., et al., Radiation therapy of pituitary tumors: results in 95 cases. Am J Clin Oncol, 1991. 14(1): p. 25-9.
- Moyes, V.J., et al., Treatment of Nelson's syndrome with temozolomide. Eur J Endocrinol,
2009. 160(1): p. 115-9.
- Losa, M., et al., Salvage therapy with temozolomide in patients with aggressive or metastatic pituitary adenomas: experience in six cases. Eur J Endocrinol, 2010. 163(6): p.
843-51.
- Salehi, F., et al., Low immunohistochemical expression of MGMT in ACTH secreting pituitary tumors of patients with Nelson syndrome. Endocr Pathol, 2010. 21(4): p. 227-9.
- Bruno, O.D., et al., Temozolomide Therapy for Aggressive Pituitary Tumors: Results in a Small Series of Patients from Argentina. Int J Endocrinol, 2015. 2015: p. 587893.
- Kelestimur, F., et al., The effects of octreotide in a patient with Nelson's syndrome. Postgrad Med J, 1996. 72(843): p. 53-4.
- Petrini, L., et al., Long-term treatment of Nelson's syndrome by octreotide: a case report. J
Endocrinol Invest, 1994. 17(2): p. 135-9.
- Daniel, E., et al., A prospective longitudinal study of Pasireotide in Nelson's syndrome Pituitary, 2018. 21(3): p. 247-255.
- Hayashi, K., et al., The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing's disease. Eur J Endocrinol, 2016. 174(2): p. 213-26.
Scarica Articolo PDF





